| Argentina, Adm. Nac. de Medicamentos, Alimentos y Tec. Médica (ANMAT) |
|
- modifica y/o complementa a: - modificada y/o complementada por: disposición 11247/16 ANMAT. |
|
Administración Nacional de Medicamentos, Alimentos y Tecnología Médica ESPECIALIDADES MEDICINALES - RIESGO SANITARIO SIGNIFICATIVO - RECOMENDACIONES- Disposición (ANMAT) 3185/99. Del 25/6/1999. B.O.: 2/7/1999. Apruébanse las recomendaciones técnicas contenidas en el documento "Cronograma para exigencias de estudios de equivalencia entre medicamentos de riesgo sanitario significativo". Bs. As., 25/6/99 VISTO la Ley 24.766, el Decreto 150/92 modificado por los Decretos 1890/92 y 177/93, la Resolución Conjunta 988/92 (M.E. y O. y S.P.) y 748/92 (M.S. y A.S.), y el Expediente Nº 1-47-11003/98-8 del registro de esta Administración Nacional y; CONSIDERANDO: Que las precitadas normas, y las disposiciones complementarias dictadas en su consecuencia, constituyen el ordenamiento legal aplicable a la aprobación, registro y autorización de venta de las especialidades medicinales cuya elaboración, importación y comercialización en el país se desarrolla al amparo de los preceptos generales establecidos por la Ley 16.463. Que quedó consolidado así un sistema fiscalizador de la actividad con el objetivo primario de garantizar que en la elaboración e importación de especialidades medicinales la eficacia, seguridad y calidad de los productos quedará plenamente certificada de acuerdo con estándares internacionales, mediante su registro ante la autoridad sanitaria nacional. Que el actual desarrollo de esos estándares torna necesaria la adopción de lineamientos técnicos que posibiliten mantener el nivel de fiscalización en los mismos parámetros internacionales con que el sistema fue configurado. Que con tal propósito corresponde adoptar para la fiscalización de especialidades medicinales en nuestro país exigencias de estudios de equivalencia, respecto de aquellos principios activos que en países de alta vigilancia sanitaria son sometidos a tales estudios y que, por su indicación terapéutica y condiciones de seguridad en el uso, deben ser consideradas como sustancias de riesgo sanitario ponderable. Que el grado de desarrollo alcanzado actualmente por el sistema fiscalizador de nuestro país incluye el diseño de los protocolos de investigación de ensayos clínicos cuyos requisitos están impuestos por la Disposición Nº 5330-ANMAT-97, encontrándose allí descriptos los procedimientos de buenas prácticas de investigación en estudios de farmacología clínica sobre los que pueden vehiculizarse las exigencias de estudios de bioequivlaencia "in vivo". Que han tomado la intervención de su competencia la comisión técnica ad-hoc conformada con representantes de las áreas técnicas del organismo y la Dirección de Asuntos Jurídicos. Que se actúa en virtud de las facultades conferidas por el Decreto 1490/92. Por ello; EL DIRECTOR NACIONAL DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA DISPONE: Artículo 1º — Apruébanse las recomendaciones técnicas para la realización de estudios de equivalencia contenidas en el documento: "Cronograma para exigencia de estudios de equivalencia entre medicamentos de riesgo sanitario significativo" cuyo texto se reproduce como Anexo I de la presente Disposición formando parte de la misma. Art. 2º — La implementación de la exigencia de estudios de equivalencia se realizará de acuerdo con el cronograma operativo incluido en el documento aprobado por el artículo precedente. Art. 4º — Regístrese; dése a la Dirección Nacional del Registro Oficial para su publicación en el Boletín Oficial. Cumplido, archívese. (Nota Ecofield: por art. 1° de la Disposición N° 108/2024 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 10/01/2024 se incorpora a la exigencia de realización de estudios de Bioequivalencia, establecidos por la presente Disposición a los ingredientes farmacéuticos (IFAs) de forma farmacéutica sólida oral que tengan como indicación la inmunosupresión en el contexto de la prevención del rechazo en el transplante de órganos y pertenezcan al grupo terapéutico de inmunosupresores, que se encuentren registrados a la fecha, o los que en un futuro se registren o incorporen en el REM, además de los que se encuentran comprendidos en un listado de exigencia de estudios de bioequivalencia in vivo o in vitro con anterioridad. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial) (Nota Ecofield: por art. 1° de la Disposición N° 107/2024 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 09/01/2024 se incorpora a la exigencia de realización de estudios de Bioequivalencia, establecidos por la presente Disposición a los ingredientes farmacéuticos (IFAs) de forma farmacéutica sólida oral que tengan como indicación la prevención o tratamiento del síndrome convulsivo en el contexto de la epilepsia y pertenezcan al grupo terapéutico de antiepilépticos, que se encuentren registrados a la fecha o los que en un futuro se inscriban e incorporen en el REM, además de los que se encuentran comprendidos en un listado de exigencia de estudios de bioequivalencia in vivo o in vitro con anterioridad. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial) (Nota Ecofield: por art. 1° de la Disposición N° 9465/2022 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 25/11/2022 se incorpora a la exigencia de realización de estudios de Bioequivalencia, establecidos por la presente Disposición a los ingredientes farmacéuticos activos VALSARTAN-SACUBITRILO, ESLICARBAZEPINA, DROSPIRENONA y FINGOLIMOD. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial) (Nota Ecofield: por art. 1° de la Disposición N° 9708/2019 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 3/12/2019 se incorpora a la exigencia de realización de estudios de Bioequivalencia, establecidos por la presente Disposición a los ingredientes farmacéuticos activos Alprazolam, Bisoprolol, Pregabalina, Memantina y Enalapril. Vigencia: a partir del día hábil siguiente al de su publicación en el Boletín Oficial) (Nota Ecofield: por art. 1° de la Disposición N° 3154/2019 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 9/4/2019 se incorpora a la exigencia de realización de estudios de Biodisponibilidad y Bioequivalencia, establecidos por la presente Disposición al Ingrediente Farmacéutico Activo IBRUTINIB. Vigencia: a partir del día hábil siguiente al de su publicación en el Boletín Oficial) (Nota Ecofield: por art. 1°
de la Disposición N° 4788/2012 de la Administración Nacional de
Medicamentos, Alimentos y Tecnología Médica B.O. 22/08/2012, se
incorpora a las exigencias de realización de estudios de
Bioequivalencia/Biodisponibilidad, establecidas por la presente
Disposición, a los ingredientes farmacéuticos activos que figuran en el
Anexo I de la norma de referencia) (Nota Ecofield: por art. 1°
de la Disposición N° 4132/2012 de la Administración Nacional de
Medicamentos, Alimentos y Tecnología Médica B.O. 24/07/2012 se incorpora
a la exigencia de demostración de bioequivalencia establecida en la
presente Disposición, a todas las concentraciones comercializadas y/o a
comercializarse de una especialidad medicinal, de forma farmacéutica
sólida oral, que contenga alguno de los Ingredientes Farmacéuticos
Activos incluidos en la normativa nacional mencionada en el VISTO de la
norma de referencia, y en disposiciones complementarias posteriores de
exigencia de bioequivalencia. Vigencia: a partir del día siguiente al de
su publicación en el Boletín Oficial) (Nota Ecofield: por art. 1° de la Disposición N° 3113/2010 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 6/7/2010 se incorporan a la exigencia de realización de estudios de Bioequivalencia / Biodisponibilidad, establecidos por la presente Disposición, a los ingredientes farmacéuticos activos Lamotrigina y Topiramato. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial) (Nota Ecofield: Por art. 1° de la Disposición N° 2446/2007 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 30/4/2007, se incorpora a la exigencia de realización de estudios de bioequivalencia / biodisponibilidad establecidos en la presente Disposición, a los principios activos Serolimus, Everolimus, Tacrolimus y Micofenolato. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial.) (Nota Ecofield: Por art. 1° de la Disposición N° 2807/2002 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 21/6/2002, se incorpora a la exigencia de realización de estudios de bioequivalencia / biodisponibilidad establecidos en la presente Disposición, al principio activo ISOTRETINOINA para uso oral. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial.) CONTENIDO CRONOGRAMA PARA EXIGENCIA DE ESTUDIOS DE EQUIVALENCIA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO II) GUIA PARA LA REALIZACION DE ESTUDIOS DE EQUIVALENCIA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO I-OBJETIVO II-DEFINICIONES Biodisponibilidad III-DEMOSTRACION DE EQUIVALENCIA TERAPEUTICA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO IV-REQUERIMIENTOS PARA LA REALIZACION DE ESTUDIOS DE EQUIVALENCIA EN SERES HUMANOS II) OBJETIVOS DEL PROYECTO I-OBJETIVO GENERAL II-OBJETIVOS INTERMEDIOS III) CRITERIOS PRELIMINARES PARA IMPLEMENTAR UN CRONOGRAMA PARA MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO QUE REQUIERAN ESTUDIOS DE EQUIVALENCIA I-CATEGORIAS DE RIESGO SANITARIO II-DROGAS ORDENADAS POR FORMA FARMACEUTICA Y POR RIESGO. III-DROGAS ORDENADAS POR EXIGENCIA, RIESGO Y VENTANA TERAPEUTICA. ANEXO I CRONOGRAMA PARA EXIGENCIA DE ESTUDIOS DE EQUIVALENCIA ENTRE MEDICAMENTOS CON ALTO RIESGO SANITARIO. CRONOGRAMA PARA EXIGENCIA DE ESTUDIOS DE EQUIVALENCIA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO. I.- GUIA PARA LA REALIZACION DE ESTUDIOS DE EQUIVALENCIA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO. I.- OBJETIVO El objetivo de esta guía es establecer recomendaciones técnicas sobre la realización de estudios de equivalencia entre medicamentos de riesgo sanitario significativo en concordancia con las pautas internacionalmente vigentes. II.- DEFINICIONES A los efectos del presente documento se adoptan las siguientes definiciones: BIODISPONIBILIDAD Es la cantidad y velocidad con las que el principio activo contenido en una forma farmacéutica alcanza la circulación sistémica, determinadas mediante la curva concentración/tiempo o la excreción urinaria. (OMS 1996). BIOEQUIVALENCIA Dos especialidades medicinales son bioequivalentes cuando siendo equivalentes farmacéuticos o alternativas farmacéuticas sus biodisponibilidades después de la administración en la misma dosis molar son semejantes en tal grado, que pueda esperarse que sus efectos sean esencialmente los mismos. (OMS 1996). EQUIVALENCIA Dos productos farmacéuticos son equivalentes cuando son farmacéuticamente equivalentes y después de administrados en la misma dosis molar sus efectos, con respecto a eficacia y seguridad, son esencialmente los mismos (MERCOSUR) (Ver EQUIVALENCIA TERAPEUTICA, OMS) EQUIVALENTE FARMACEUTICO Dos especialidades medicinales son equivalentes farmacéuticos si contienen la misma cantidad de principio activo, en la misma forma farmacéutica, están destinados a ser administrados por la misma vía y cumplen con estándares de calidad idénticos o comparables. Sin embargo, la equivalencia farmacéutica no necesariamente implica equivalencia terapéutica ya que diferencias en los excipientes, en el proceso de elaboración, u otras pueden determinar disparidades en el comportamiento de los productos. (OMS). EQUIVALENCIA TERAPEUTICA Dos especialidades medicinales son equivalentes terapéuticos cuando siendo alternativas o equivalentes farmacéuticos y después de la administración en la misma dosis molar sus efectos con respecto a la eficacia y seguridad resultan esencialmente los mismos, luego de estudios apropiados (de bioequivalencia, farmacodinámicos, clínicos o in-vitro). (OMS). ALTERNATIVA FARMACEUTICA Productos que dentro del concepto de producto similar: a) Contiene el mismo principio terapéutico, siendo diferente la salificación, esterificación o complejación del mismo, o b) Se presentan en diferentes formas farmacéuticas o concentraciones por unidad de administración, poseyendo la misma vía de administración, la misma indicación terapéutica y la misma posología (CPM, 12/1998). PRODUCTO DE REFERENCIA Producto para el cual la eficacia y seguridad han sido establecidas. Cuando el producto innovador no se encuentre disponible, el líder del mercado puede ser utilizado como producto de referencia (OMS, 1996), o el que determine la autoridad sanitaria para cada caso. PRODUCTO SIMILAR A los fines de la presente norma se entiende como tal al producto que contiene la(s) misma(s) sustancia(s) terapéuticamente activas como base de su formulación, así como formas farmacéuticas, vías de administración, posología, indicaciones, contraindicaciones, precauciones, advertencias, reacciones adversas, pruebas de disolución y otros datos correlativos semejantes al producto registrado en el país o países de los Anexos correspondientes, pudiendo diferir en características tales como tamaño y forma, excipientes, período de vida útil, envase primario. III.- DEMOSTRACION DE EQUIVALENCIA TERAPEUTICA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO 1.- GENERALIDADES Una vez cumplimentadas las buenas prácticas de manufactura y control de calidad, para la demostración de equivalencia terapéutica los métodos y muestras experimentales considerados por orden decreciente de seguridad, sensibilidad y reproducibilidad son: 1.1. Estudios de bioequivalencia (Estudios Farmacocinéticos): 1.1.1. Determinación de la concentración del principio activo o sus metabolitos en sangre total, plasma, suero o en otro fluido biológico en función del tiempo o; 1.1.2. Determinación de la excreción urinaria del principio activo o sus metabolitos en función del tiempo. 1.2. Estudios farmacodinámicos "agudos" 1.2.1. Determinación de un efecto farmacológico agudo producido por el producto problema y de referencia o sus metabolitos en función del tiempo si dicho efecto puede ser determinado con suficiente seguridad, sensibilidad y reproducibilidad. Esta metodología puede ser aplicable cuando no existan métodos disponibles para la determinación de los productos problema y de referencia en los fluidos biológicos o excretorios. 1.3. Ensayos clínicos comparativos. 1.4. Ensayos de disolución "in vitro". 1.4.1. Prueba/s de disolución u otras, correlacionadas o no con estudios de biodisponibilidad, pero que se hallan descriptas en las farmacopeas reconocidas internacionalmente o en la bibliografía científica con evidencias suficientes de validación. 1.4.2. Prueba/s realizadas mediante métodos (que produzcan resultados correlacionados con estudios en seres humanos, para medicamentos con riesgo sanitario significativo), con evidencias suficientes de validación. 1.4.3.- Indicadores básicos: Tiempo de disolución de una determinada cantidad de principio activo; cantidad de principio activo disuelto en un tiempo determinado, en medio o medios que se hallen correlacionados con el empleo terapéutico del producto terminado. 2.- APLICACION DE LOS ESTUDIOS DE EQUIVALENCIA ENTRE MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO 2.1.- NO REQUIEREN ESTUDIOS DE EQUIVALENCIA 2.1.1. Productos destinados a ser administrados parenteralmente (por ejemplo: vías intravenosa, intramuscular o intratecal) en soluciones acussas que contengan el/los mismo/s principio/s activo/s en la/s misma/s concentración/es (OMS, 1996). 2.1.2. Soluciones para utilización por vía oral que contengan el/los mismo/s principios/s activo/s en la /s misma/s concentraciones y no contengan un excipiente que se conozca o se sospeche que afecta el tránsito gastrointestinal o la absorción del principio activo (OMS, 1996). 2.1.3. Gases o vapores (Res. 3784/91; OMS, 1996). 2.1.4. Polvos y/o granulados para reconstitución como solución, cuando cumpla con los puntos 2.1.1. y 2.1.2. 2.1.5. Productos otológicos u oftalmológicos que contengan el/los mismo/s principio/s activo/s en la/s misma/s concentración/es y esencialmente los mismos excipientes (OMS, 1996). 2.1.6. Productos para empleo tópico, líquidos, que contengan el/los mismos/s principio/s activo/s y esencialmente el/los mismo/s excipiente/s (OMS,1996). 2.1.7. Formas farmacéuticas de aplicación tópica (crema, pomada, gel, etc.), de uso externo, sólidas, que contengan el/los mismos/s principio/s activo/s y esencialmente el/los mismo/s excipiente/s. 2.1.8. Productos destinados a ser utilizados por inhalación o aerosoles nasales que sean administrados con o sin esencialmente el mismo dispositivo, sean preparados como soluciones acuosas y que contengan el/los mismos/s principio/s activo/s y esencialmente el/los mismo/s excipiente/s en concentraciones comparables (OMS, 1996). 2.1.9. Productos conteniendo principios activos de administración oral que no deban absorberse (Res. 3784/91). Se considera "Esencialmente los mismos excipientes" a excipientes del mismo tipo en cuanto a que posean la misma función en la formulación (dispersante, agregante, espesante, etc.), aunque no se trate de la misma molécula. 3.- REQUIEREN SOLAMENTE ESTUDIOS DE EQUIVALENCIA "IN-VITRO" 3.1. Comprimido de liberación simple. 3.2. Cápsula de liberación simple. 3.3. Diferentes concentraciones de un producto cuando: 3.3.1.-La composición cualitativa de las diferentes concentraciones es esencialmente la misma; 3.3.2.-La relación principio activo-excipiente, para las diferentes concentraciones es esencialmente la misma o para concentraciones bajas la relación entre los excipientes es la misma; 3.3.3.- Se ha realizado un estudio apropiado de equivalencia para al menos una de las concentraciones de la formulación (usualmente la concentración mayor, a menos que se haya elegido la concentración menor por razones de seguridad); 3.3.4.-En el caso que la disponibilidad sistémica, haya demostrado una farmacocinética lineal dentro del rango terapéutico. De acuerdo a normas de la Unión Europea, serán excepción a los estudios "In-vivo" cuando se haya demostrado una aceptable correlación entre la tasa de disolución "In-vivo" e "In-vitro" y la tasa de disolución "In-vitro" del nuevo medicamento sea equivalente a la del medicamento ya autorizado, en las mismas condiciones de prueba utilizadas para establecer la correlación. 4.- REQUIEREN ESTUDIOS DE EQUIVALENCIA "IN-VITRO" E "IN VIVO" (de bioequivalencia, farmacodinámico o ensayo clínico controlado). Para los medicamentos con riesgo sanitario significativo se procederá a la realización de estudios "In-vivo" en las siguientes situaciones: 4.1.-Formas farmacéuticas de liberación modificada controlada, sostenida , programada, etc. 4.2. Sistemas terapéuticos. 4.3.- Formas farmacéuticas de liberación simple que contengan principios activos que reúnan una o más de las siguientes características: 4.3.1.- Propiedades fisicoquímicas desfavorables: - Escasa solubilidad en agua (menor a 0,1%). - Variaciones cristalográficas metaestables. - Baja humectabilidad. 4.3.2- Características farmacocinéticas: - Farmacocinética No Lineal en todo el rango terapéutico (de orden 0, no proporcional o dosis dependiente). - Escasa tasa de absorción (menor del 30%). - Estrecha ventana terapéutica : definiéndose la misma como: a) El cociente entre la Dosis letal media (DL50) y la Dosis eficaz media (DE50), es menor de 2. b) El cociente entre la concentración tóxica mínima y la concentración eficaz mínima es menor de 2. c) El uso eficaz y seguro de las especialidades que contienen la droga en cuestión, requiere cuidadosa dosificación y monitoreo del paciente. - Elevado metabolismo de primer pasaje hepático (mayor del 70%). 4.3.3.- Características farmacodinámicas - Curva Dosis-Respuesta "empinada" (es decir, pequeños cambios en la dosis determinan impor-tantes variaciones en los efectos). - Estrecho margen de seguridad (cociente DL50/DE50, menor a 2). 4.3.4.- Características clínicas - Evidencia clínica de problemas relacionados con la biodisponibilidad. IV.- REQUERIMIENTOS PARA LA REALIZACION DE ESTUDIOS DE EQUIVALENCIA EN SERES HUMANOS. 1.-GENERALIDADES De acuerdo a lo establecido en la Disposición 5330/97 (Ensayos Clínicos), o la que en su caso la reemplace, en lo que se refiere a: -Ambito de aplicación y de sus alcances. -Autorización, seguimiento y controles del estudio. -Requisitos de los investigadores y de los patrocinantes. -Incumplimiento de la normativa. -Los requisitos básicos. -Los centros en donde se llevará a cabo el estudio. -Los requerimientos éticos. 2.- INFORMACION SOBRE LOS PRODUCTOS UTILIZADOS EN EL ESTUDIO. 2.1. Las características farmacocinéticas/dinámicas del producto de referencia y la forma farmacéutica a utilizar en el ensayo y del producto problema si las hubiera. No será imprescindible la presentación de información preclínica para los estudios de bioequivalencia (Disp. 5330/97, Capítulo VII, Introducción). 2.2. La composición cualicuantitativa del producto problema. El contenido de droga activa del producto problema con respecto al producto de referencia no debe diferir en más de un 5%. Si ello ocurriera, esta diferencia debe ser tenida en cuenta en el cálculo de los datos de biodisponibilidad para facilitar la comparación de ambos productos farmacéuticos. 2.3. Los test de disolución "in vitro" comparativos entre el lote del producto de referencia y problema que serán utilizados en el ensayo o el test "in vitro" que corresponda según la forma farmacéutica a utilizar durante el ensayo. 2.4. Drogas 2.4.1. Productos de referencia y en estudio: Nombre aprobado. Nombre comercial. Fabricante. Forma farmacéutica. Dosis. Número de lote. 2.4.2. Provisión, almacenamiento y dispensación. 2.4.3. Acondicionamiento, etiquetado y aleatorización. 2.5. (Punto derogado por art.
5° de la Disposición N° 1263/2012 de la Administración Nacional de
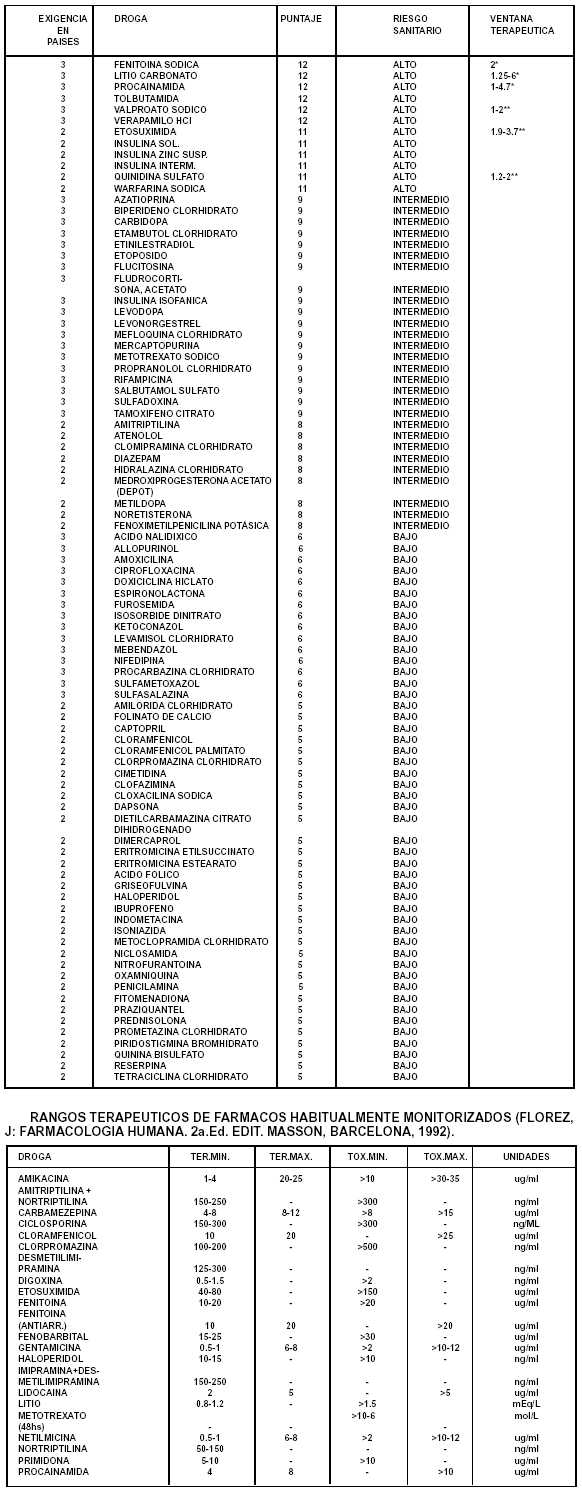
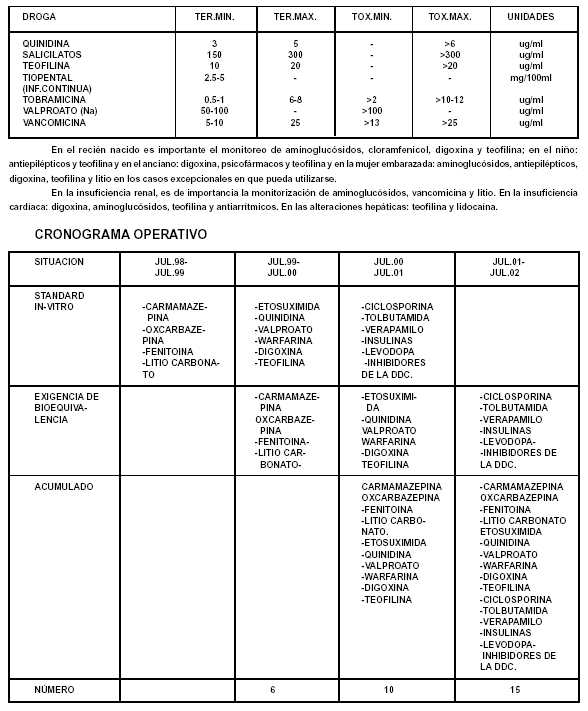
Medicamentos, Alimentos y Tecnología Médica B.O. 8/3/2012) 2.6. Reserva de muestras: se debe guardar cantidad suficiente de cada lote bajo condiciones de almacenamiento apropiadas de los productos farmacéuticos utilizados de los estudios de equivalencia en seres humanos, así como sus protocolos de análisis y características. Estas muestras podrán ser requeridas por las autoridades en caso de ser necesario. 3.- SUJETOS 3.1. Ensayo de bioequivalencia: Los participantes deberán ser preferiblemente voluntarios sanos de ambos sexos entre 18 a 55 años de edad y de peso acorde a los rangos aceptados. No deben tener antecedentes de abuso de drogas o alcohol y ser no-fumadores. Se establecerán criterios de inclusión / exclusión, de acuerdo a las características de los productos en estudio. Se confeccionarán historias clínicas detalladas las cuales incluirán: exámenes físico y de laboratorio. Si el estudio fuera realizado en pacientes esta alternativa deberá ser justificada. Los participantes serán monitoreados durante y con posterioridad al desarrollo del estudio con el objeto de tomar las medidas adecuadas en caso de aparición de reacciones adversas, toxicidad o alguna enfermedad intercurrente. Tamaño de la muestra: De 18 a 24 voluntarios sanos y en ningún caso menor de 12. El número de sujetos participantes deberá ser justificado. 3.2. Ensayo farmacodinámico: Seguirán los mismos lineamientos que para los ensayos de bioequivalencia anteriormente descriptos y podrán participar tanto voluntarios sanos como pacientes. 3.3. Ensayo clínico comparativo: Seguirán los lineamientos establecidos en la Disposición 5330/97, o la que en su caso la reemplace, siendo habitualmente el número de pacientes participantes mucho mayor a los requeridos para los estudios de bioequivalencia El número de sujetos participantes deberá justificarse. 4.- PRODUCTO DE REFERENCIA Y EN ESTUDIO 4.1. Producto de referencia (Ver II.- DEFINICIONES). 4.2. Producto problema: Es la alternativa farmacéutica o el equivalente farmacéutico del producto de referencia. 5.- METODOS ANALITICOS. 5.1.- Ensayo de bioequivalencia. Los métodos analíticos usados para determinar el principio activo y/o sus productos de biotransformación en plasma, suero, sangre u orina u otro fluido biológico deberán cumplimentar los requerimientos de especificidad, sensibilidad, precisión y seguridad. Se deberán informar los resultados de validación de los procedimientos a utilizar en el ensayo (p.e.: curva standard, límite de detección del principio activo y/o de su producto de biotransformación). Se deberán informar los resultados de los ensayos de estabilidad del principio activo y/o de su producto de biotransformación en la muestra experimental. 5.2.- Ensayo farmacodinámico. La metodología utilizada para determinar el efecto farmacológico o terapéutico relevante deberá cumplimentar los requerimientos de especificidad, sensibilidad, precisión y seguridad. Dichos métodos incluyen: registros de los eventos mediante instrumental adecuado o escalas analógicas visuales o categorización cualitativa de los datos. 5.3.- Ensayo clínico comparativo. La metodología utilizada comprenderá todos aquellos procedimientos clínicos adecuados que permitan determinar el comienzo y la intensidad del efecto terapéutico relevante según Disposición 5330/97 o la que en su caso la reemplace. 6.- DISEÑO EXPERIMENTAL 6.1. Dosis única del producto problema y de referencia: 6.1.1.Generalidades: - Realizado en voluntarios sanos que recibirán una única dosis tanto del producto problema como de referencia. -El producto problema y de referencia deberá ser administrado a sujetos en ayunas, a menos que otro método puede ser más apropiado el cual deberá justificarse. -El diseño será cruzado, a menos que un diseño paralelo o de otro tipo resulte más apropiado el cual deberá justificarse. -A menos que otras razones lo justifiquen el período de eliminación deberá ser determinado durante: a- Por lo menos tres veces la vida media del principio activo o su metabolito en sangre o en orina; o b- Por lo menos tres veces la vida media de desaparición del efecto farmacológico agudo. 6.1.2. Según la muestra experimental: -Sangre a- La colección de las muestras sanguíneas serán realizadas con la suficiente frecuencia para permitir la determinación: a.1. De la concentración máxima. a.2. El área bajo la curva por un periodo de tiempo de por lo menos tres veces la vida media del principio activo o su metabolito. b- Según la forma farmacéutica: b.1. En un estudio en que se comparen las formas orales del producto problema y de referencia la periodicidad de la colección de las muestras debe ser idéntica. - Orina Cuando la comparación del producto problema y de referencia se basa en la determinación de una curva acumulativa de excreción urinaria en función de tiempo la colección de las muestras de orina deberá realizarse con la suficiente frecuencia para permitir la determinación de la velocidad y cantidad excretada de la droga o sus metabolismos. 6.2. Efecto farmacológico agudo 6.2.1. Cuando la comparación del producto problema y de referencia se basa en la determinación de un efecto farmacológico agudo en función del tiempo, las determinaciones de estos efectos deberán ser realizadas con la suficiente frecuencia para permitir una razonable estimación del área bajo la curva por un periodo de tiempo de por lo menos tres veces la vida media de desaparición del efecto farmacológico, la respuesta máxima y el tiempo en que esa respuesta máxima se manifiesta. 6.2.2. El uso de un efecto farmacológico agudo para la determinación de la equivalencia puede requerir la realización de una curva dosis-respuesta, en tales casos la equivalencia puede ser determinada por comparación de la curva dosis-respuesta como así también del área bajo la curva en función del tiempo para una dosis determinada. 6.3. Dosis múltiples del producto problema y de referencia: 6.3.1. Generalidades En determinadas circunstancias la comparación del producto problema y de referencia requerirá la administración repetida de los mismos con el objeto de obtener niveles estacionarios del principio activo en el organismo. 6.3.2. Condiciones para la realización de ensayos de bioequivalencia con dosis múltiple en medicamentos con riesgo sanitario significativo. Se requerirá un estudio empleando dosis múltiples cuando: - Existan diferencias en la velocidad de absorción pero no en la cantidad absorbida. - Haya una excesiva variabilidad en la biodisponibilidad entre sujetos. - La concentración del principio activo o sus metabolismos en sangre resultante de la administración de una única dosis se encuentran por debajo de los límites de detección del método analítico. - El producto problema es una forma de liberación controlada. - Farmacocinética dosis o tiempo-dependiente. 6.3.3. Diseño experimental - Será realizado en voluntarios sanos que recibirán dosis múltiples tanto del producto problema como de referencia. - El producto problema y de referencia deberá ser administrado a sujetos en ayunas, a menos que otro método pueda ser más apropiado el cual deberá justificarse. - El producto problema y de referencia deberá ser administrado de acuerdo a las recomendaciones usuales de posología. - El diseño será cruzado, a menos que un diseño paralelo o de otro tipo resulte más apropiado por razones científicas válidas y deberá prever un período adecuado de eliminación de la droga si las condiciones de equilibrio no se alcanzan. - A menos que otras razones lo justifiquen el período de eliminación deberá ser determinado durante: a- Por lo menos cinco veces la vida media del principio activo o su metabolito en sangre o en orina; o b- Por lo menos cinco veces la vida media de desaparición del efecto farmacológico agudo. 6.3.4. Según la muestra experimental -Para el ensayo de dosis múltiple: a-Sangre y Orina: Cuando la comparación entre la droga problema y de referencia se base en curvas de concentración sanguínea o en curvas acumulativas de excreción urinaria en función del tiempo en estado de equilibrio las muestras de sangre u orina deberán ser tomadas con la suficiente frecuencia durante dos o más días consecutivos para definir adecuadamente la concentración plástica o urinaria máxima y mínima en equilibrio. -Para el efecto farmacológico agudo: Cuando la comparación entre la droga problema y de referencia se base en la determinación de una curva efecto-tiempo las mediciones de dicho efecto serán lo suficientemente frecuentes para demostrar un efecto máximo y ausencia de diferencias significativas entre ambos productos. 7.- VARIABLES FARMACOCINETICAS: Según el diseño experimental y las muestras biológicas: Cmax, Cmaxx, Cmaxr, Cmin, Cp, Tmax, ABCt, ABC¥, ABCt, ABCx, ABCr TMR, Aet, Ae¥, dAe / dt, t 1/2. Cmax: Concentración plasmática máxima. Cmaxx: Concentración plasmática máxima del producto problema. Cmaxr: Concentración plasmática máxima del producto de referencia. Cmin: Concentración plasmática mínima. Cp : Concentración plasmática promedio. Tmax: tiempo en el cual se alcanza la Cmax. ABCt: área bajo la curva de concentración plasmática en un tiempo t. ABC¥: área bajo la curva de concentración plasmática extrapolada al infinito. ABCt: área bajo la curva de concentración plasmática durante un intervalo de dosis en estado deequilibrio. ABCx: área bajo la curva del producto problema ABCr: área bajo la curva del producto de referencia TMR: tiempo medio de residencia. Aet: excreción urinaria acumulativa a un tiempo t. Ae¥: excreción urinaria acumulativa extrapolada al infinito. dAe / dt: velocidad de excreción urinaria. t1/2: vida media de concentración plasmática. 8.- ANALISIS ESTADISTICO. Serán aceptados aquellos procedimientos estadísticos que no excedan el nivel de significancia del 5% y entre ellos aquellos con el menor riesgo de rechazar erróneamente equivalencia. Podrán ser utilizados métodos paramétricos o no paramétricos según corresponda. 9.- CRITERIOS DE EQUIVALENCIA: 9.1. Para Bioequivalencia: ABCx / ABCr: El intervalo de confianza 90% de la razón entre ambas ABC deberá estar contenido dentro de rango de bioequivalencia de 0.8 - 1.25. Cmax x / Cmax r : Debido a la variabilidad de este parámetro los rangos serán más amplios que para la razón ABC, la elección del rango deberá ser justificada. El cálculo de cualquier otra variable utilizada para declarar dos productos bioequivalentes deberá ser justificada. 9.2. Para ensayo farmacodinámico agudo: Debe tenerse en cuenta que los intervalos utilizados en los ensayos de bioequivalencia son habitualmente muy amplios como para ser aplicados a este tipo de ensayos, por esta razón deberán ser definidos caso por caso y descriptos en el protocolo. 9.3. Para ensayos clínicos comparativos De acuerdo a lo establecido en la Disposición 5330/97 o la que en su caso la reemplace. II.- OBJETIVOS DEL PROYECTO I.- OBJETIVO GENERAL: Establecer un cronograma para la exigencia progresiva de estudios de bioequivalencia para medicamentos o especialidades medicinales similares. II.- OBJETIVOS INTERMEDIOS: 1. Fijar criterios de selección de principios activos a los cuales se les exigirá estudios de bioequivalencia. 2. Establecer standard "In-vitro" para los principios activos seleccionados, necesariamente previos a la exigencia de estudios de Bioequivalencia. El establecimiento del objetivo 2.2. encuentra su fundamentación en el hecho que es necesario establecer un standard "In-vitro", previo a la realización de estudios "In-vivo", a los efectos de controlar las variables dependientes del medicamento, previas a la administración "In-vivo". III.-CRITERIOS PRELIMINARES PARA IMPLEMENTAR UN CRONOGRAMA PARA MEDICAMENTOS CON RIESGO SANITARIO SIGNIFICATIVO QUE REQUIERAN ESTUDIOS DE EQUIVALENCIA. Para esta aproximación se partió del documento: ‘‘WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS." WHO Technical Report Series Nº 863, Geneva,1996, del cual se extrajo de la tabla 1, pág. 142: "Examples of national requierments studies". En primer lugar se procedió a ensillar las drogas que Canadá, Alemania y EE.UU. coinciden en que deben ser sometidas a estudios de bioequivalencia, es decir, que las tres autoridades sanitarias están contestes en que deben ser estudiadas in-vivo (categorización +b de la tabla referida). Posteriormente se clasificaron las drogas de acuerdo al Riesgo Sanitario, tomando para la fijación del mismo, dos aspectos: a.- Terapéutico: principios activos utilizados en desórdenes serios ya sea porque ponen en peligro la vida (por ejemplo algunos antiarrítmicos) o porque poseen complicaciones graves (por ejemplo algunos antiepilépticos). b.- Seguridad: principios activos que poseen una estrecha relación entre su concentración máxima efectiva no tóxica y su concentración mínima efectiva (por ejemplo las sales de litio o los preparados de teofilina). Luego se enlistaron las drogas con las que poseen acuerdo 2 de los países de alta vigilancia sanitaria (EE.UU., Canadá y Alemania), y dentro de estos principios activos, también se procedió a subclasificarlos de acuerdo al Riesgo Sanitario. CATEGORIAS DE RIESGO SANITARIO RIESGO SANITARIO ALTO: Es la probabilidad de aparición de complicaciones de la enfermedad amenazantes para la vida o para la integridad psicofísica de la persona y/o de reacciones adversas graves (OMS) cuando la concentración sanguínea de la droga no se encuentra dentro de la ventana terapéutica. RIESGO SANITARIO INTERMEDIO: Es la probabilidad de aparición de complicaciones de la enfermedad no amenazantes para la vida o para la integridad psicofísica de la persona y/o de reacciones adversas no necesariamente graves cuando la concentración sanguínea de la droga no se encuentra dentro de la ventana terapéutica. RIESGO SANITARIO BAJO: Es la probabilidad de aparición de una complicación menor de la enfermedad y/o de reacciones adversas leves cuando la concentración sanguínea de la droga no se encuentra dentro de la ventana terapéutica. Operativamente se asignó un puntaje ponderado, consistente en lo siguiente: (RIESGO X 3) + (EXIGENCIA PAIS X 1 ) RIESGO ALTO: 3 PUNTOS RIESGO INTERMEDIO: 2 PUNTOS RIESGO BAJO: 1 PUNTO EXIGENCIA EN 3 PAISES: 3 PUNTOS EXIGENCIA EN 2 PAISES: 2 PUNTOS
REFERENCIAS BIBLIOGRAFICAS (1)- CFR. 320.1. 4.01.90. (2)- FDA Home Page (Internet). 28.08.96. (3)- Goodman Gilman, L. Las bases farmacológicas de la terapéutica 8 va Ed. Médica Panamericana. Bs. As. 1991. (4)- OMS-OPS: Glosario de términos especializados para la evaluación de medicamentos. Washington, 1990. (5)- OMS. Consultative document. Interchangeable multi-source pharmaceutical products. 06.12.93. (6)- WHOth.Technical Report Series Nº 863. Expert Committee on Specifications for pharmaceutical preparations. 34 th Report. Geneva,1996. BIBLIOGRAFIA CONSULTADA - Cid Cárcamo, E.: Introducción a la Farmacocinética. Monografía Nº 25. Serie Billoogía. Secretaría General de la Organización de los Estados Americanos. Programa Regional de Desarrollo Científico y Tecnológico. Washington, 1982. - Cid Cárcamo, E.: La cinética de disolución de medicamentos. Monografía Nº 24. Serie Billoogía. Secretaría General de la Organización de los Estados Americanos. Programa Regional de Desarrollo Científico y Tecnológico. Washington, 1981. - EEC Guidelines investigation of bioavalability and bioequivalence: Rules governing medicinal products in the European Community. Vol III, 159-161(Cit. 5), 1990. - FDA: Aprove drug products with therapeutic equivalence evaluation- 11th De., 1991. -Flórez, J: Farmacología humana. 2a.Ed. Edit. Masson, Barcelona, 1992. - Glasser, AC: Aspects for bioavailability and bioequivalence revision. Possible implications on clinical pharmacology. Meth. Find. Exptl. Clin. Pharmacol. 1987, 9,4 49-452 (Cit. 5). -Goodman Gilman’s Las bases farmacológicas de la terapéutica. 9a. Ed. Interamericana. México, 1996. - Greenblat, DJ: et al.: Bioequivalence of generic drugs in clinical psychopharmacology. J. Clin. Psychopharmacology 1987, A21-A23 (Cit. 5). - Iannantuono, R; Tessler, J: Biodisponibilidad y Bioequivalencia. Revista Argentina de Farmacología Clínica 1994,1,5,226-243. - Koch-Wesser, J: Bioavailability of drugs (first of two parts). N. Engl. J. Med. 1974,291,233-237. - Marzo, A.: Bioequivalence. Drugs made Germany. 1995,38,1,6-13. - Noticias Farmacéuticas 1989,10,16-17. -Steinijans, VW et al.: Controversies in bioequivalence studies. Clin. Pharmacokin. 1992, 22, 4,247- 253. - Stromb, BL: Generic Drug Substitution revisited. N. Engl. J. Med. 1987,316, 1456-1462. - WHO: Draft guidelines on marketing authorization requierements. Interchangeable multisarcepharmaceutical products. Consultative document. December 6, 1993.
|
|
-o- |